Método Monte Carlo Cinético para o Cálculo do Equilíbrio de Adsorção Langmuiriana de Espécies Gasosas em Superfícies Cristalinas
Autores
- Ana Luísa A. Alves (Graduação - IC - IBiotec - Ciências Biológicas)
- Luis Vinicius C. Silva (Pós-graduando - PG - PPGCEM - Doutorado em Ciência e Engenharia de Materiais)
- Email: luisvinicius@ufcat.edu.br
Afilição: Universidade Federal de Catalão
Palavras-chave
- Adsorção Langmuir
- Monte Carlo Cinético
- Simulação
- Físico-Química
Introdução
- A simulação de adsorção é fundamental para diversas aplicações como o desenvolvimento de catalisadores e técnicas de tratamento de efluentes [3].
- Este estudo apresenta um Método Monte Carlo Cinético (MMCC) para calcular a evolução temporal da taxa de ocupação dos sítios de uma superfície cristalina adsorvendo um gás, de acordo com o modelo de Langmuir.
- Melhoria no Algoritmo: Aplicação de uma malha adaptativa para minimizar a variância da solução numérica.
Suposições do modelo:
- Moléculas adsorvidas não interagem significativamente entre si.
- Cada molécula ocupa um único sítio de adsorção em uma superfície homogênea.
- O equilíbrio é atingido quando as taxas de adsorção (rA) e desorção (rD) se igualam.
Metodologia
- Resolução inicial e máxima:
- Malha inicial: 32x32
- Malha máxima: 256x256
- Tempo real máximo (t): 10 unidades.
- Algoritmo simplificado do MMCC ilustrado na Figura 1.
Resultados e Discussões
- Erros menores que 3% foram observados em todas as simulações.
- Resolução máxima necessária: 128x128 para a maioria dos cenários; 256x256 para os dois últimos.
- A Figura 2 compara o estado final do lattice e a solução obtida pelo MMCC com a solução analítica no Cenário 1.
Cenários simulados e resultados:
| Cenário | TA (sítios/tempo) | TD (sítios/tempo) | Erro Relativo (Original) | Erro Relativo (Modificado) | Diferença de Tempo (s) |
|---|---|---|---|---|---|
| 1 | 0.5 | 1.0 | 1.88% | 1.92% | -1.2 |
| 2 | 0.5 | 2.0 | 1.85% | 1.81% | -0.5 |
| 3 | 2.0 | 1.0 | 2.58% | 2.49% | -1.8 |
| 4 | 1.0 | 1.0 | 0.86% | 0.80% | -1.9 |
| 5 | 1e-5 | 1.0 | 2.86% | 2.83% | 1.6 |
| 6 | 1.0 | 1e-5 | 1.92% | 1.92% | -0.4 |
Experimentos
Dois experimentos foram realizados para ilustrar a dinâmica de adsorção simulada pelo MMCC:
Simulação com ( T_a = 1 ) e ( T_d = 1 ):
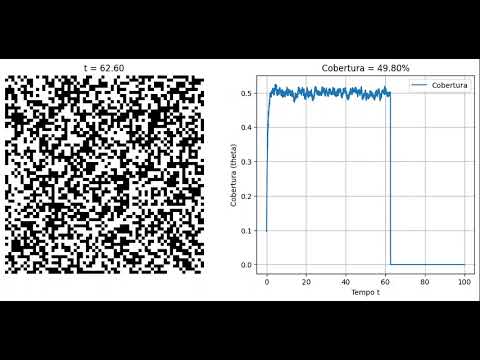
Título: Kinetic Monte Carlo simulating Langmuir adsorption modelSimulação com ( T_a = 2 ) e ( T_d = 1 ):
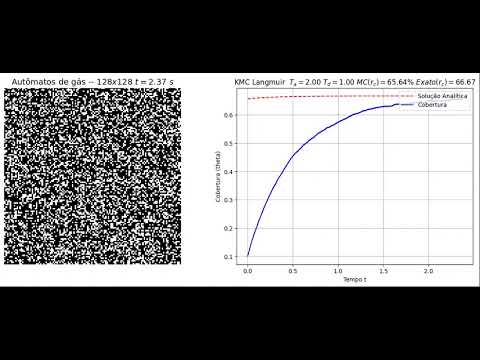
Título: Kinetic Monte Carlo simulating Langmuir adsorption model
Conclusões
- Dinâmicas de adsorção e desorção podem ser modeladas como um processo de Poisson e simuladas por MMCC.
- Malhas adaptativas resultaram em convergência mais rápida e maior acurácia.
- Estudos futuros podem incluir:
- Obtenção de taxas específicas (adsorção/desorção) usando técnicas experimentais assim como computacionais, e.g: DFT e MD [2].
- Simulação de sistemas complexos como filmes finos, materiais porosos e catalisadores com geometrias variadas.
Material adicional
Códigos estão disponíveis no repositório GitHub do meu doutorado, sinta-se livre para modificar e citar esse trabalho.
Um demo no Unity pode ser rodado via browser aqui
O app para Android da simulação pode ser baixado aqui
O painel em PDF pode ser baixado aqui
Agradecimentos
- Luis Vinicius C. Silva agradece o suporte financeiro fornecido pela FAPEG, essencial para o avanço desta pesquisa.
Referências
- Fichthom, E.; Weinberg, K. A. Theoretical Foundations of Dynamical Monte Carlo Simulations. J. Chem. Phys. 1991, 95, 1090–1096.
- Daigle, A. D.; BelBruno, J. J. Density Functional Theory Study of the Adsorption of Nitrogen and Sulfur Atoms on Gold Surfaces. J. Phys. Chem. C 2011, 115 (46), 22987–22997.
- Bahamon, D.; Vega, L. F. Pharmaceutical Removal from Water Effluents by Adsorption on Activated Carbons: A Monte Carlo Simulation Study. Langmuir 2017, 33 (42), 11146–11155.